Displaying items by tag: Theory
A fresh perspective for organic monolayers on silicon: new paper
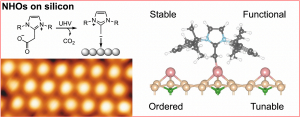
The controlled formation of stable ordered monolayers of N-heterocyclic olefins on a surface has been demonstrated for the first time. This exciting new multidisciplinary work, performed by CNR-ISM theoreticians in collaboration with Italian (University of Rome "Tor Vergata") and German (University of Münster and Technical University of Berlin) surface physicists and organic chemists, has been published in Angewandte Chemie.
Ab-initio Many-Body Methods and Simulations with the Yambo Code - An ICTP school for PhD and Researchers

The Computational School on Ab-initio Many-Body Methods and Simulations with the Yambo Code will introduce many-body perturbation theory (MBPT) approaches and specifically to first-principles excited-state simulations using the YAMBO. code.
The target participants are graduate students, postdocs, and researchers who are interested in learning or in improving their knowledge and skills to calculate electronic and optical properties of an efficient, beyond the well-known DFT limitations and using an efficient, highly parallelized and accurate many-body computational tool.
#ab_initio, #yambo_code, #school, #computational_physics, #condensed_matter, #manybody_perturbation_theory, #quasiparticles, #excitons, #highperformancecomputing, #gpu
Selective Effects of the Host Matrix in Hydrogenated InGaAsN Alloys: Toward an Integrated Matrix/Defect Engineering Paradigm
Article published on Advanced Functional Material.
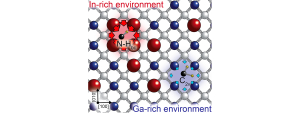
In dilute nitride InyGa1−yAs1−xNx alloys, a spatially controlled tuning of the energy gap can be realized by combining the introduction of N atoms—inducing a significant reduction of this parameter—with that of hydrogen atoms, which neutralize the effect of N. In these alloys, hydrogen forms N–H complexes in both Ga-rich and In-rich N environments.
Here, photoluminescence measurements and thermal annealing treatments show that, surprisingly, N neutralization by H is significantly inhibited when the number of In-N bonds increases. Density functional theory calculations account for this result and reveal an original, physical phenomenon.
#dilute_nitride_alloys #density_functional_theory
BOOSTER - Boosting sustainability in plastic electronics: the key role of functional surfactants as reaction medium and dispersing agents
Development of new, highly sustainable strategies for the synthesis of organic materials for electronic and opto-electronic devices.
EFOR II - Energy from renewable sources
The project aimed to understand, through the construction of ES systems with different configurations, if and under which conditions the coupling between hard and soft phases allows to improve the thermal stability compared to systems consisting of the hard phase only.
EFOR I - Energy from Renewable Sources
The project aimed to understand, through the construction of ES systems with different configurations, if and under which conditions the coupling between hard and soft phases allows to improve the thermal stability compared to systems consisting of the hard phase only.
POLYPHEMO - POLymer based hYbrid nanomaterials for PHotovoltaics: improving Efficiency by theoretical Modeling
Development of a multiscale theoretical method for the investigation of semiconductor/molecule/polymer interfaces playing a key role in bulk-heterojunction solar cells.
BIOX - Predicting and controlling the fate of bio-molecules driven by extreme-ultraviolet radiation
The target of the project is the investigation, with extreme temporal resolution, of the molecular processes initiated by the interaction of ionizing radiation with biologically relevant molecules.
EOS - Organic Electronics for Innovative Research Instrumentation
Development of new organic materials for the realization of electronic devices and integrated circuits for scientific applications.
MultiscaleChemBio
Development of a multiscale theoretical method for the investigation of photosynthetic natural and artificial systems.
 English (UK)
English (UK)  Italiano (Italia)
Italiano (Italia)