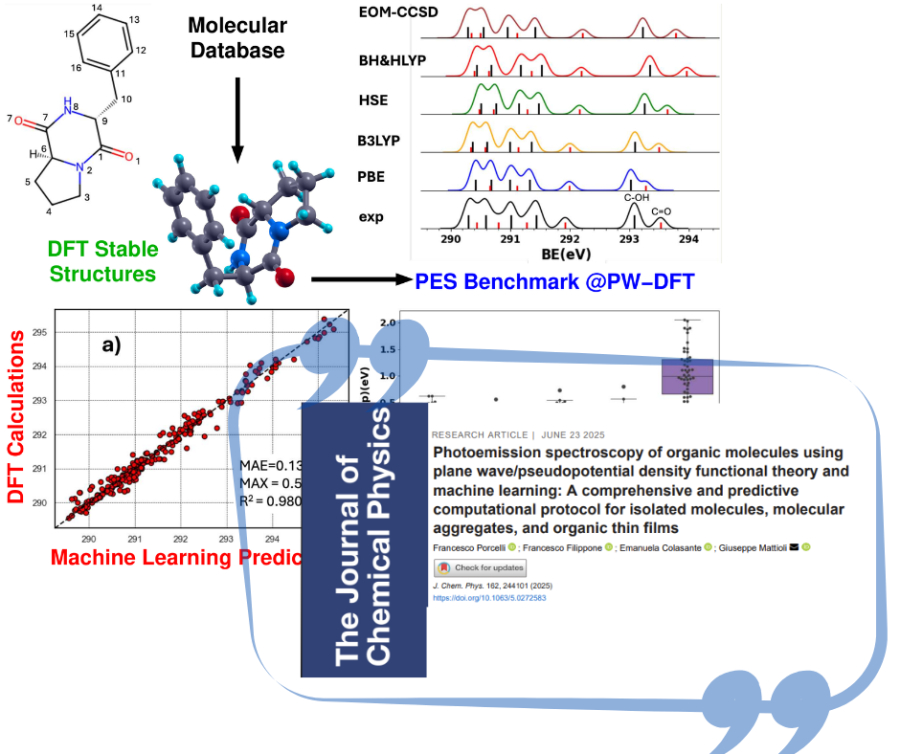
We have developed and validated a robust computational protocol based on plane wave density functional theory (PW-DFT) and a ΔSCF approach for predicting X-ray photoemission spectra (XPS) of isolated molecules. Our method enables accurate calculation of core-level binding energies (BEs), specifically for C1s, N1s, and O1s orbitals, by incorporating core-hole pseudopotentials and assessing various exchange–correlation functionals (PBE, B3LYP, HSE, BH&HLYP). Our extensive benchmarking, including comparison with equation-of-motion coupled-cluster (EOM-CCSD) calculations and experimental data, demonstrated strong agreement across a broad set of molecular classes—aromatic, aliphatic, heteroaromatic compounds, drugs, and biomolecules. Each functional's strengths and weaknesses were evaluated: for example, PBE offers low-cost calculations but tends to underestimate core BEs in polar environments, while B3LYP and HSE showed consistent accuracy across different chemical environments. We extended our protocol's application to large molecular aggregates and thin films on inorganic surfaces, showing promising results for modeling realistic systems such as N-doped graphite and surface-adsorbed molecular layers. In valence photoemission, Kohn–Sham eigenvalues were tested as proxies for ionization potentials across diverse molecules, with BH&HLYP showing the best accuracy and transferability. Importantly, we have also developed an initial machine learning (ML) model trained on PW-DFT data to predict XPS spectra of organic molecules. The model effectively reproduced experimental spectral features, highlighting the growing role of ML in spectroscopic prediction. Future developments will incorporate conformational and intermolecular interaction effects to improve prediction fidelity. To support reproducibility and community adoption, we provide an open repository with core-hole pseudopotentials, input files, and training datasets for ML applications.
This work has been financially supported by ICSC-Centro Nazionale di Ricerca in High Performance Computing, Big Data, and Quantum Computing, funded by European Union-NextGenerationEU (Grant No. CN00000013), and by the Italian Minister of the University and Research (MUR) within the PRIN-2022 research program (project “NIR+,” Grant No. 2022BREBFN)
 English (UK)
English (UK)  Italiano (Italia)
Italiano (Italia)