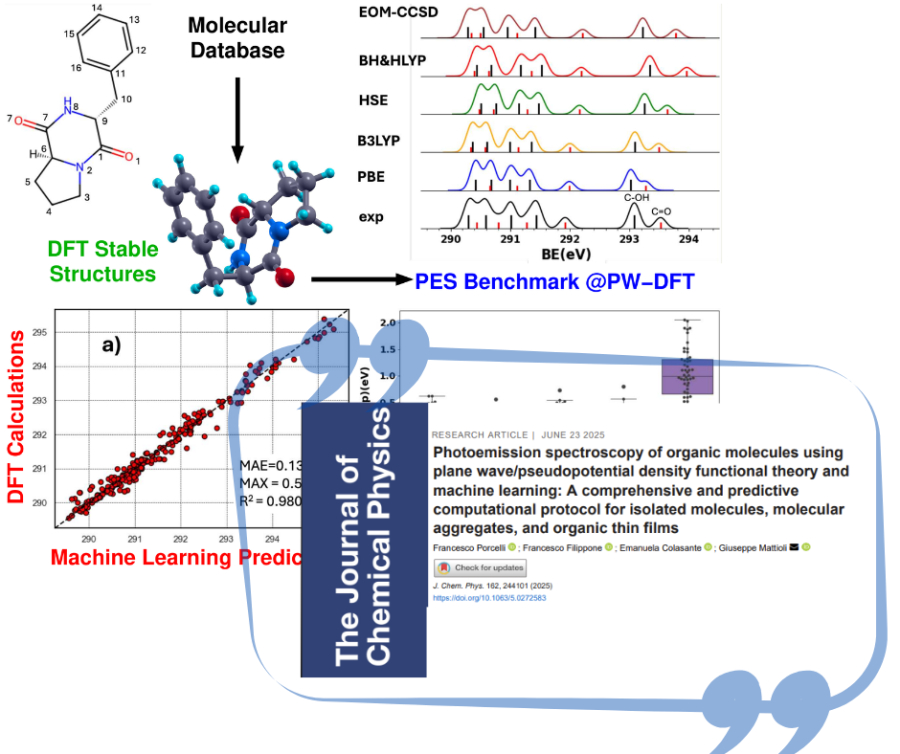
Abbiamo sviluppato e validato un protocollo computazionale solido basato sulla teoria del funzionale della densità a onde piane (PW-DFT) e su un approccio ΔSCF per la previsione degli spettri di fotoemissione X (XPS) di molecole isolate. Il nostro metodo consente il calcolo accurato delle energie di legame (BE) dei livelli core C1s, N1s e O1s, utilizzando pseudopotenziali con lacuna nel core e valutando diversi funzionali di scambio–correlazione (PBE, B3LYP, HSE, BH&HLYP). Il protocollo è stato ampiamente testato e confrontato con dati sperimentali e calcoli EOM-CCSD, mostrando un’eccellente accuratezza su un ampio spettro di molecole: aromatiche, alifatiche, eteroaromatiche, farmaci e biomolecole. Abbiamo analizzato i punti di forza e le debolezze di ciascun funzionale: PBE è efficiente ma sottostima le BE in ambienti polari, mentre B3LYP e HSE garantiscono una buona accuratezza in diversi contesti chimici. Abbiamo inoltre esteso l’applicabilità del protocollo a grandi aggregati molecolari e film sottili su substrati inorganici, ottenendo risultati promettenti per sistemi realistici come la grafite drogata con azoto e superfici funzionalizzate. Per la fotoemissione di valenza, abbiamo valutato gli autovalori di Kohn–Sham come stime delle potenziali di ionizzazione, riscontrando che BH&HLYP offre la migliore accuratezza e trasferibilità tra i sistemi esaminati. Inoltre, abbiamo sviluppato un modello preliminare di machine learning (ML) addestrato su dati PW-DFT per la previsione degli spettri XPS di molecole organiche, dimostrando una buona capacità di riprodurre le caratteristiche spettrali sperimentali. Prevediamo di migliorare il modello ML includendo effetti conformazionali e interazioni intermolecolari per aumentarne l’affidabilità. Per favorire la riproducibilità e la diffusione del metodo, mettiamo a disposizione un repository pubblico con pseudopotenziali, file di input e dataset per l’addestramento dei modelli ML.
Questo lavoro è stato finanziato da ICSC–Centro Nazionale di Ricerca in High Performance Computing, Big Data e Quantum Computing, con il supporto dell’Unione Europea–NextGenerationEU (Grant n. CN00000013), e dal Ministero dell’Università e della Ricerca (MUR) nell’ambito del programma di ricerca PRIN-2022 (progetto “NIR+”, Grant n. 2022BREBFN).
 Italiano (Italia)
Italiano (Italia)  English (UK)
English (UK)